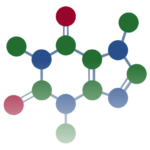
量子化学計算・分子力場計算では、ある分子の3次元構造を出発点として様々な計算を実行します。
この記事では、計算の材料となる分子の3次元構造をどのように作成するかを紹介します。
・xyzファイル、PDBファイル、MOL2ファイルの例
・計算化学を始めるためにおすすめのソフトウェア(無償・有償)
・計算実行までの簡単な流れ
計算化学でよく使う構造データ
3次元構造データは様々なものがあり、迷ってしまうこともあるかと思います。
量子化学計算・分子力場計算では以下の3つ作成できるようになっていれば基本的には大丈夫です。
アミノ酸のアラニンの3次元構造を例に、実際のデータを見ておきましょう。
xyzファイル
その名の通り、各原子のCartesian座標(x, y, z座標)が記載されているデータです。
原子間のつながり(結合次数等)の情報を必要としない量子化学計算では、このデータで十分です。
拡張子は.xyzです。
↓実際のデータ
13 N -0.96600 0.49300 1.50000 C 0.25700 0.41800 0.69200 C -0.09400 0.01700 -0.71600 O -1.05600 -0.68200 -0.92300 C 1.20400 -0.62000 1.29600 O 0.66100 0.43900 -1.74200 H -1.38300 -0.42500 1.48200 H -0.67600 0.66100 2.45200 H 0.74600 1.39200 0.68200 H 1.45900 -0.33000 2.31600 H 0.71500 -1.59400 1.30700 H 2.11300 -0.67600 0.69700 H 0.43500 0.18200 -2.64700
PDBファイル
タンパク質の構造データベースであるProtein Data Bank (PDB)でダウンロードできる形式です。
原子の種類(同じ炭素でも脂肪族炭素なのか芳香族炭素なのかといった違いなど)や、どの原子同士が結合しているかといった情報が含まれているため、分子力場計算や分子動力学シミュレーションではこちらを使うことが多いです。
タンパク質のデータベースに基づいているため、アミノ酸や核酸などの生体分子については、その残基レベルで詳細な記載が可能です。
拡張子は.pdbです。
↓実際のデータ
COMPND UNNAMED AUTHOR ATOM 1 N ALA A 1 -0.966 0.493 1.500 1.00 0.00 N ATOM 2 CA ALA A 1 0.257 0.418 0.692 1.00 0.00 C ATOM 3 C ALA A 1 -0.094 0.017 -0.716 1.00 0.00 C ATOM 4 O ALA A 1 -1.056 -0.682 -0.923 1.00 0.00 O ATOM 5 CB ALA A 1 1.204 -0.620 1.296 1.00 0.00 C ATOM 6 OXT ALA A 1 0.661 0.439 -1.742 1.00 0.00 O ATOM 7 H1 ALA A 1 -1.383 -0.425 1.482 1.00 0.00 H ATOM 8 H2 ALA A 1 -0.676 0.661 2.452 1.00 0.00 H ATOM 9 HA ALA A 1 0.746 1.392 0.682 1.00 0.00 H ATOM 10 HB1 ALA A 1 1.459 -0.330 2.316 1.00 0.00 H ATOM 11 HB2 ALA A 1 0.715 -1.594 1.307 1.00 0.00 H ATOM 12 HB3 ALA A 1 2.113 -0.676 0.697 1.00 0.00 H ATOM 13 HXT ALA A 1 0.435 0.182 -2.647 1.00 0.00 H CONECT 1 2 7 8 CONECT 2 1 3 5 9 CONECT 3 2 4 6 CONECT 4 3 CONECT 5 2 10 11 12 CONECT 6 3 13 CONECT 7 1 CONECT 8 1 CONECT 9 2 CONECT 10 5 CONECT 11 5 CONECT 12 5 CONECT 13 6 MASTER 0 0 0 0 0 0 0 0 13 0 13 0 END
MOL2ファイル
PDBと同様に結合次数等の情報を含んだ分子の3次元構造ファイルです。
PDBファイルのように残基の情報は含まないため、xyzファイルとPDBファイルの中間のようなイメージのデータです。
拡張子は.mol2です。
↓実際のデータ
@<TRIPOS>MOLECULE
*****
13 12 0 0 0
SMALL
GASTEIGER
@<TRIPOS>ATOM
1 N -0.9660 0.4930 1.5000 N.3 1 ALA1 -0.3186
2 CA 0.2570 0.4180 0.6920 C.3 1 ALA1 0.1004
3 C -0.0940 0.0170 -0.7160 C.2 1 ALA1 0.3214
4 O -1.0560 -0.6820 -0.9230 O.2 1 ALA1 -0.2493
5 CB 1.2040 -0.6200 1.2960 C.3 1 ALA1 -0.0395
6 OXT 0.6610 0.4390 -1.7420 O.3 1 ALA1 -0.4795
7 H1 -1.3830 -0.4250 1.4820 H 1 ALA1 0.1189
8 H2 -0.6760 0.6610 2.4520 H 1 ALA1 0.1189
9 HA 0.7460 1.3920 0.6820 H 1 ALA1 0.0571
10 HB1 1.4590 -0.3300 2.3160 H 1 ALA1 0.0250
11 HB2 0.7150 -1.5940 1.3070 H 1 ALA1 0.0250
12 HB3 2.1130 -0.6760 0.6970 H 1 ALA1 0.0250
13 HXT 0.4350 0.1820 -2.6470 H 1 ALA1 0.2951
@<TRIPOS>BOND
1 1 2 1
2 1 7 1
3 1 8 1
4 2 3 1
5 2 5 1
6 2 9 1
7 3 4 2
8 3 6 1
9 5 10 1
10 5 11 1
11 5 12 1
12 6 13 1
構造データを作成するためのソフトウェア
3次元構造データを作成するためのソフトウェアは様々なものがあります。
ここでは代表的なものをいくつか紹介しますが、それぞれ使用感が異なりますので、好みのものを探してみてください。
テキストエディター
構造データや計算の実行ファイルを扱うためにはテキストでの処理が必要です。
Windows標準のメモ帳等でもできないことはないですが、テキストを見やすく表示したり、テキスト内の検索や置換をスムーズに行うために他のものをインストールしておくとよいでしょう。
どれがよいのかは好みになりますが、以下のものが人気のようです。
・Visual Studio Code
・Atom (管理人も使っていましたが、開発終了になってしまいました。。。)
・Notepad++
ChemDraw・Chem3D
ChemDrawは多くの方が使用されていると思いますが、Chem3DはそのChemDrawと連携したソフトウェアです。
ChemDrawと同時にインストールされていることも多く、ChemDrawで描画した2次元構造式から簡単に3次元構造データを作成できるのでおすすめです。
Avogadro
Avogadroは、化学を学ぶ学生や研究者向けの無料で使えるオープンソースの分子描画ソフトです。使いやすい操作性が特徴で、分子モデルの作成や編集、視覚化が簡単に行えます。
Avogadro2も開発されていますが、オリジナル版の方が正直使いやすいです。
使い方は以下のサイトで紹介されています。
Avogadroをつかってみよう
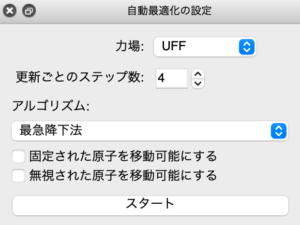
一から3Dで作成した構造はいびつになりがちですが、簡易的な分子力場計算による自動最適化の機能が搭載されているのでこれを利用するときれいな構造をつくることができます。自動最適化のウィンドウは下の画像のようになっています。スタートを押せば構造最適化が走ります。
PyMOL
PyMOLは、生命化学の分野でよく使われる分子視覚化ソフトウェアです。タンパク質や小さな分子の構造を3Dで美しく描くことができるので、論文やプレゼン資料の作成に適しています。
使用方法については
おうちでできる構造解析(旧大阪大学蛋白質研究所)
PyMOLの表示形式プリセット集(Qiita @Ag_smith (Yoshitaka Moriwaki))
等のサイトでわかりやすくまとめられています。
主に構造の確認に用いることが多いですが、PDBファイル→xyzファイル等、ファイル形式の変換は可能となっています。
オープンソース版もあり、この導入については
Qiita: オープンソース版PyMOLインストール (Windows)@hnishi
Qiita: macOS/Ubuntu 22.04へのオープンソース版PyMOL 3.0のインストール方法 @Ag_smith (Yoshitaka Moriwaki)
で詳しく紹介されています。
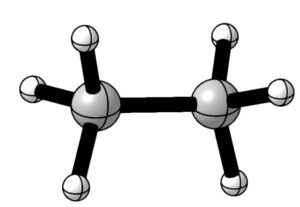
CYLview20
こちらも無償のソフトウェアでGaussianの計算結果を開いて構造を表示することができます。
エネルギーの推移等も分子の構造とともに見ることができるので、GaussViewは使えないけどGaussianで計算したいときに重宝します。
Viewタブから設定できるスタイルのうち、「Houkmol」を選択すると、以下の画像のように論文でよく見るデザインの図を作成することができます。
Chemcraft
Chemcraftは、構造を作成するためだけではなく、多くの量子化学計算プログラムの計算結果を視覚化し、分析するための機能が備わっています。
有償のプログラムですが、お値段に見合った機能がたくさん付いているので予算的な問題をクリアできるのであれば導入を検討してみる価値ありです。
GaussView
こちらも有償のプログラム。
GaussViewは量子化学計算プログラムのGawssian専用ソフトといってもよいでしょう。
非常に高価ですが、計算のインプット作成をGUIで行うことができ、結果の解析も簡単に行えます。
計算開始までの流れ
最後に計算までの大まかな流れを書いておきます。
①計算したい分子の構造を上記プログラムを活用して作成する
ChemDrawからChem3Dにもっていったり、結晶構造データをPyMOLでxyz形式やPDB形式に変換したり。
AvogadroやChemcraftで直接3次元構造を作成してもよいです。
たいていのプログラムでは、ファイルの保存かエクスポートでPDBファイル、xyzファイル、MOL2ファイルとして保存することができます。
②インプットファイルを作成する
各計算プログラムごとのルールに従い、インプットファイル(計算実行のための仕様書のようなもの)を作成します。
(こちらについてはプログラムごとの個別に記事にします。)
GaussView等を用いていると3次元構造をGUI上で見ながら作成するイメージになると思いますが、完全にテキストベースで作成することができます。
様々な計算オプションを組み込むためにはテキストで編集できることがほぼ必須です。
慣れてきたら直接テキストファイルで作成できるようにしましょう。
③インプットファイルをプログラムに渡して計算を実行する
これについても個別に記事とする予定です。
実行方法も、一つ一つの計算を個別に実行することや、複数を連続して処理する方法などがあります。
終わりに
この記事を参考に、様々な分子の3次元構造データを作成してみてください。
肝心の計算のインプットファイルや計算の実行については別記事での公開を予定しています。